And Via Foundry Enhances Bulk RNA‑Seq’s Modern Potential
Emerging technologies like spatial transcriptomics, multi-omic fusion, and AI agents are creating serious buzz – and deservedly so. They're empowering researchers to ask entirely new questions, uncover deeper insights, and accelerate discovery like never before.
Today, though, we want to spotlight something that hasn’t gotten as much recent hype: Bulk RNA-seq. It might not be grabbing headlines, but it deserves real credit for its proven power, versatility, and continued impact on therapeutic development. Like a fine wine, bulk RNA-seq just keeps getting better with age.
RNA-seq’s reach spans the entire drug development process – from the first hints of a target to the final patient-response readout. Yet its very power breeds complexity. Even a proven method like bulk RNA-seq can pose significant challenges, especially without tools specifically designed to simplify its complexity.
Before we dive into how Via Foundry dissolves those roadblocks, let’s take a tour of the therapy-development pipeline to see exactly where, and why, RNA-seq makes the difference.
RNA-seq is leveraged throughout the entire therapeutic development pipeline...from initial R&D stages through clinical applications.
RNA-seq provides critical insights throughout therapeutic development, enabling researchers to dissect disease mechanisms, test treatments at the molecular level, optimize therapeutic efficacy and safety, and track responses from cell lines to human populations.
While the questions evolve at each stage of the drug development process, RNA-seq – especially through differential expression analysis – remains a critical and powerful tool for evaluating how therapies impact the transcriptome. It helps researchers identify targets, optimize drug candidates, assess safety, and understand mechanisms of action.
Early Drug Discovery & Screening
High-throughput screening enables rapid identification of promising candidate molecules. Initial assays (like qPCR or luciferase) flag potential hits, but RNA-seq takes it deeper – offering a genome-wide view of each candidate’s effects. This comprehensive perspective helps the researcher spot intended targets and potential off-target impacts early.
Target Identification and Validation
After initial hits are verified, RNA-seq gives researchers a genome-wide perspective, pinpointing candidates that effectively modulate disease-related genes while minimizing unwanted effects.
Early in the discovery phase, RNA-seq compares diseased and healthy tissues, highlighting dysregulated RNAs as potential therapeutic targets. For instance, sequencing diseased versus healthy liver tissue can reveal pathogenic RNAs – guiding targeted therapeutic strategies at both the RNA and DNA levels.
Proof of Concept
At this stage, RNA-seq confirms whether candidate therapies – like targeted RNA molecules – are driving the intended gene-expression changes in cell-line models. By quantifying transcriptomic shifts after intervention, RNA-seq provides molecular-level validation of therapeutic efficacy, ensuring confidence before moving forward.
Lead Optimization
With a lead compound in hand, RNA-seq plays a key role in guiding refinements. It helps track on-target effects – confirming whether tweaks enhance efficacy – and flags off-target impacts to ensure safety isn’t compromised. This molecular feedback loop is essential for fine-tuning both effectiveness and tolerability.
Preclinical Work
RNA-seq continues to deliver value in preclinical studies – helping clarify mechanisms of action, dose responses, and toxicity profiles. By applying different drug doses across time points and measuring transcriptomic changes, researchers can:
- Confirm target engagement and specificity
- Detect toxicity or off-target gene expression
- Assess RNA durability and persistence
These insights start in cell lines and are later validated in animal models, ensuring the therapy is well understood before clinical trials begin.
Safety Assessment
As development progresses toward clinical trials, RNA-seq becomes a key tool for safety assessment. By surveying the full transcriptome, researchers can spot unintended biological effects early and work to minimize off-target risks – ensuring greater confidence in therapeutic safety.
Clinical and Post-Clinical
As therapies move into clinical and population-level studies, RNA-seq continues to prove its value. It enables:
- Measurement of therapeutic impact in real patients
- Detection of side effects and gene expression variability across subpopulations
- Identification of biomarkers for patient stratification
Even without deep clinical detail, RNA-seq’s comprehensive view supports a better understanding of drug mechanisms, resistance, and long-term effects – while scaling to meet the demands of larger trials.
–
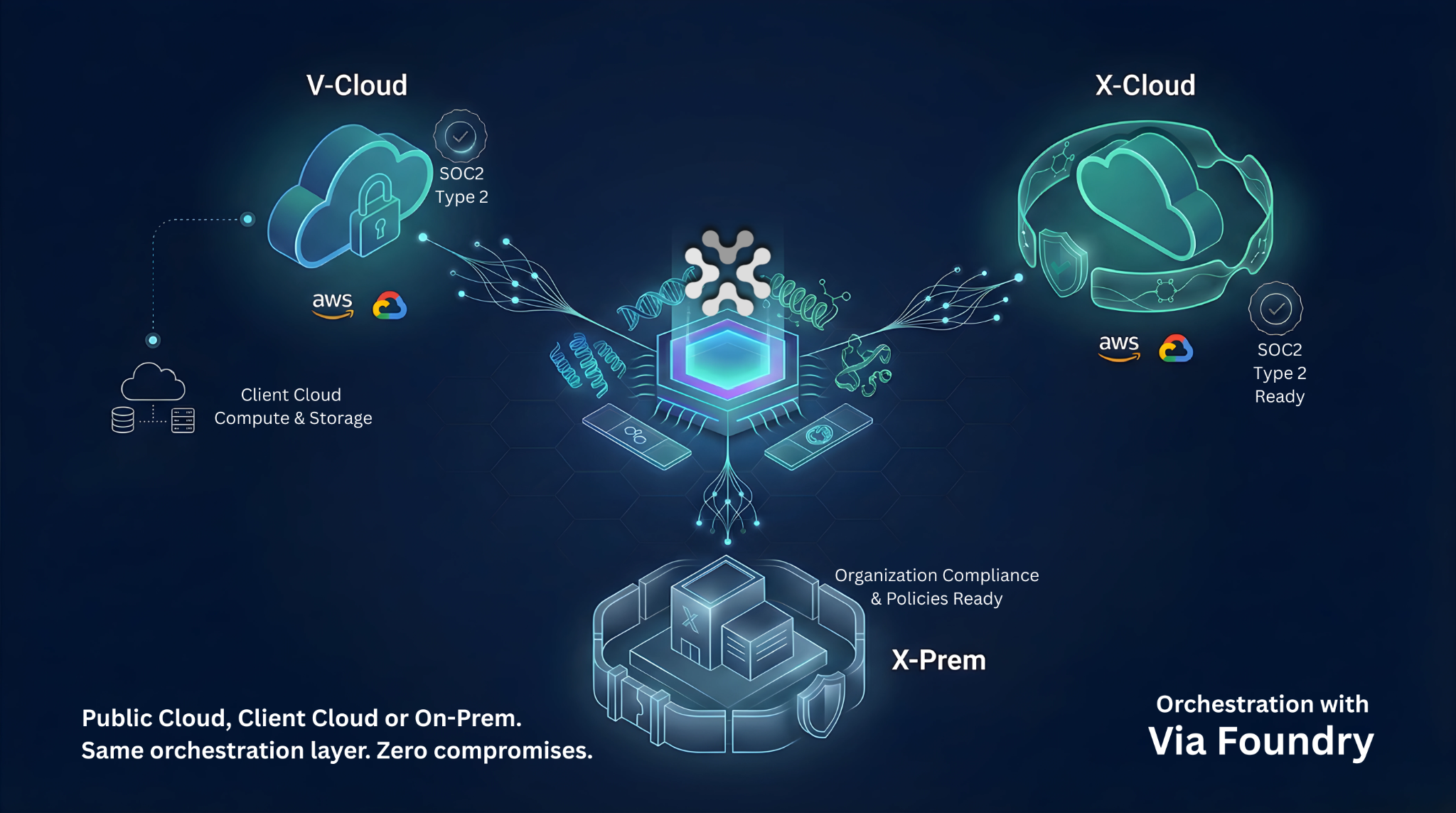
RNA-seq isn’t just a supporting assay – it’s the molecular heartbeat of modern drug development, pulsing through target discovery, lead refinement, preclinical safety screens, and every patient cohort thereafter. But its indispensable reach comes with painstaking library prep, batch effects, and multi-terabyte data hurdles that can stall even elite bioinformatics teams. Via Foundry turns that complexity into click-through clarity – abstracting the low-level grind while surfacing the signals that matter – so scientists can follow the biology, not the bottlenecks, and advance therapies with confidence.
How Via Foundry Brings Out The Best In RNA-Seq
Via Foundry streamlines RNA-seq analysis, solving core challenges of scalability and reproducibility. A single integrated pipeline takes you seamlessly through pre-processing QC, processing, and post-processing analysis – delivering analysis-ready data so you can move directly into downstream interpretation.
The video below demonstrates how Via Foundry streamlines RNA-seq analysis across the drug discovery pipeline, highlighting its modular architecture, automated workflow, and interactive data exploration capabilities.
Via Foundry delivers a seamless, end-to-end RNA-seq workflow: raw-read QC, adapter trimming, alignment, quantification, normalization, and biological interpretation all run in a single automated pipeline. It natively profiles every major RNA class – mRNA, miRNA, lncRNA, circRNA, and other small RNAs – so researchers can explore both well-established expression signatures and emerging therapeutic targets without stitching together separate tools. The result is a streamlined, deeply detailed analysis that turns complex multi-step processing into a single, intuitive experience.

Via Foundry is a reimagined version of Dolphinnext - outlined in this paper: Yukselen, O., et al. (2020). DolphinNext: a distributed data processing platform for high throughput genomics. BMC Genomics, 21, 310. https://doi.org/10.1186/s12864-020-6714-x
1. Pre-processing (Data Preparation)
This initial phase of the RNA-seq pipeline takes one or several FastQ input files. This phase is crucial for ensuring the integrity and accuracy of downstream analyses by systematically removing poor-quality sequences and technical artifacts from the raw data. It consists of several important read quality control and filtering actions (see image above):
- Read quality reports: The pipeline generates reports on the quality of the input reads. This is achieved using tools like FastQC.
- Read quality filtering: Low-quality reads can be eliminated based on user-specified criteria and thresholds. This is done using tools like trimmomatic.
- Read quality trimming: Bases at the ends of reads with low quality scores can be removed. The pipeline performs 5′ and 3′ trimming.
- Adapter removal: Sequences originating from library preparation adapters are removed to ensure accurate downstream analysis. This is done using cutadapt.
- Optional alignment and filtering of specific genomic loci: The pipeline offers the user the option to align, filter out, and/or estimate the abundance of both standard and predefined sets of genomic loci (e.g., rRNAs, miRNAs, tRNAs, piRNAs, snoRNAs, ERCC, mobile elements). This can involve alignment using Bowtie2.
- Read trimming for optimal alignment: Before any alignment, reads may be trimmed to the desired length for optimal alignment, especially for specific types of RNA like miRNAs or tRNAs or if quality issues are suspected at the read ends.
Only reads that pass all filters in the data preparation stage are kept for later steps. After this phase, the pipeline produces quality reports including FastQC reports and information about the fraction of reads aligned to each of the genomic loci selected by the user.
2. Processing (Alignment and Quantification)
This central phase involves aligning the filtered reads and quantifying the abundance of transcripts or genes. This step transforms cleaned reads into biologically meaningful insights by accurately assigning reads to transcripts or genomic regions. The RNA-seq pipeline offers flexibility in its approach:
- Transcriptome Alignment and Quantification (for expression level estimation): To estimate expression levels, the pipeline uses RSEM which aligns reads to a predefined set of transcripts. Users can choose from available transcript sets (i.e., Ensemble, GENCODE, RefSeq) or upload their own. RSEM not only aligns the reads but also provides estimated counts and transcripts per million (TPM).
- Genome Alignment (for visual assessment and quality control): The pipeline also aligns reads against the genome using splicing-aware alignment algorithms to generate a genome browser viewable file to visually assess genomic contamination and library quality. Users can choose between any, or all, of the most commonly used aligners e.g. STAR, Hisat2 and Tophat2.
3. Post-processing (Quality Evaluation and File Generation)
This phase focuses on evaluating the quality of the processing steps and generating output files for further analysis and visualization. It ensures the reliability of your data by providing clear metrics and visualization-ready files that simplify quality assessment and interpretation:
- Quality Metrics from Genomic Alignments: If the user opted to perform genomic alignments, the pipeline reports overall quality metrics such as coverage and the number of mapped reads to different genomic and transcriptomic regions (Figure S4). These reports rely on Picard’s CollectRNASeqMetrics program (http://broadinstitute.github.io/picard) and the RSeQC program.
- Genome Browser File Generation: Resulting genomic alignments are then processed to generate genome browser-friendly formats: bigwig (for UCSC genome browser) or TDF (for the Integrative Genome Viewer (IGV)). These files enable visual inspection of read coverage patterns.
4. Downstream Analysis (Quantification Output and Further Exploration)
The final phase involves the output of quantification data and options for further exploration. With data ready for biological discovery, researchers can now seamlessly transition from raw counts to meaningful biological insights through intuitive tools designed for interactive exploration:
- Quantification Matrix: The RNA-seq pipeline returns a quantification matrix that includes the estimated counts and transcripts per million (TPM) based on RSEM or by simply counting reads using featureCounts for each gene and/or for each annotated isoform.
- Interactive Data Exploration: These matrices are used as inputs for differential gene expression analysis and can be uploaded directly to an embedded instance of DEBrowser software, which allows interactive exploration of the resulting data.
Via Foundry: Reproducible, Rapid, Next-Level RNA-Seq
Bulk RNA-seq may no longer dominate the headlines, yet it remains a versatile and critical tool in modern biology.
Via Foundry harnesses that power, wrapping QC, alignment, quantification, and interpretation into one pipeline – so data moves from sequencer to insight without detours. Built-in autoscaling, versioned workflows, and automated provenance make reproducibility and compliance the default, not a chore.
The result: hours instead of days to surface differential signatures, off-target effects, or patient-stratification markers, whether you’re refining leads at the bench or orchestrating multi-site clinical trials. With Via Foundry, the trusted workhorse of RNA-seq becomes a high-performance engine propelling the next wave of discoveries.